Software on GitHub
All of our recent software is available on GitHub. Published software is highlighted below. These and other packages are all available at our Github page.
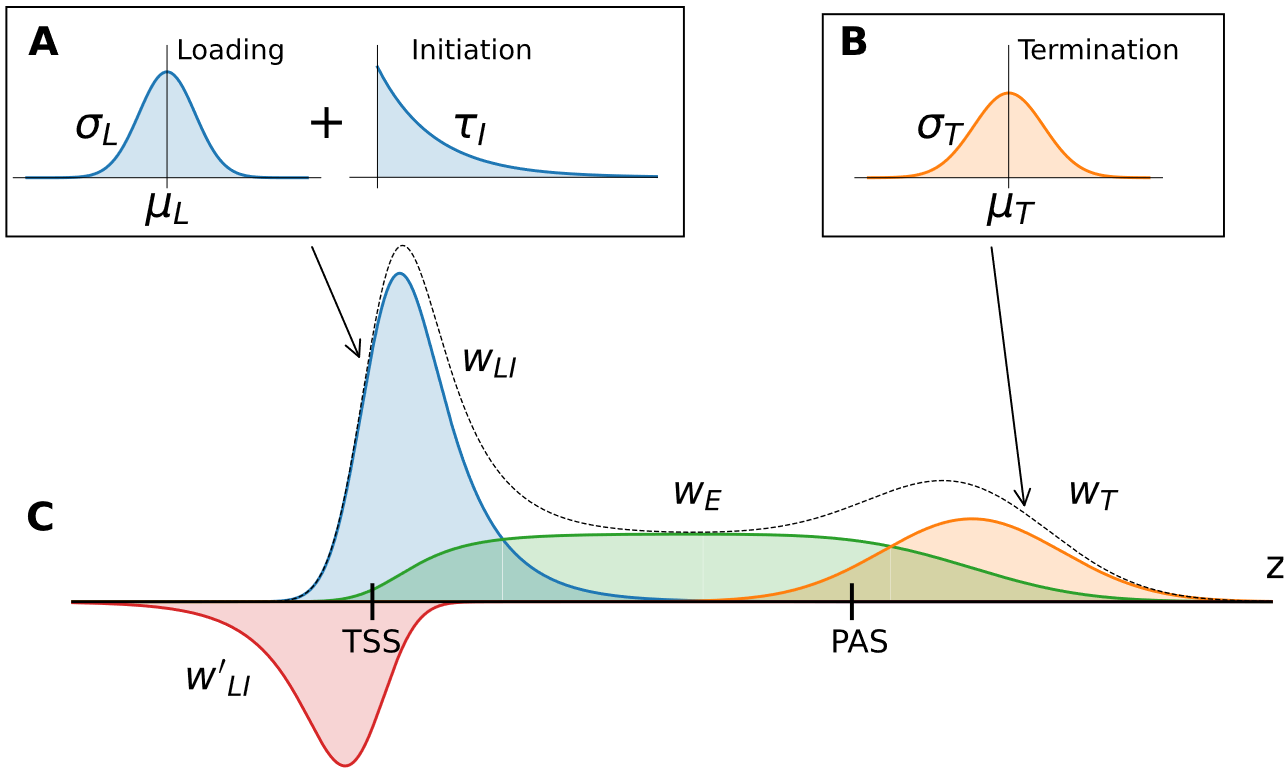
LIET models RNA polymerase activity by parameterizing each stage of transcription (Loading, Initiation, Elongation, and Termination)
Source code available for download:
GitHub
Originally published in:
J. Stanley, G.E.F. Barone, H.A. Townsend, R.F. Sigauke, M.A. Allen, R.D. Dowell
bioRxiv (2024)

TFEA performs enrichment analysis of transcription factor motifs within specific genomic regions
Source code available for download:
GitHub
Originally published in:
J.D. Rubin, J.T. Stanley, R.F. Sigauke, C.B. Levandowski, Z.L. Maas, J. Westfall, D.J. Taatjes & R.D. Dowell
Communications Biology 4:661 (2021)

Transcription fit (Tfit) utilizes a generative model of RNA polymerase to identify sites of divergent transcription in nascent transcription assays.
Source code available for download:
GitHub
Originally published in:
J.G. Azofeifa and R.D. Dowell
Bioinformatics 33(2):227-234 (2017)

FStitch is a fast and simple algorithm to detect nascent RNA transcription in global nuclear run on sequencing (GRO-seq).
Source code available for download:
GitHub
Originally published in:
J. Azofeifa, M.A. Allen, M.Lladser, and R.D. Dowell
IEEE/ACM Transactions on Computational Biology and Bioinformatics 14(5):1070-1081 (2017)
An HMM to infer the ancestral origin of distinct regions within both the ILS and ISS genomes.
Source code available for download:
GitHub
Originally published in:
R.D. Dowell, A. Odell, P. Richmond, D. Malmer, E. Halper-Stromberg, B. Bennett, C. Larson, S. Leach, R.A. Radcliffe
Mammalian Genome 27:574 (2016)
LADMA (Levenshtein Automata based Degenerate Motif Finder) accepts a number of mismatches and builds a Levenshtein non-deterministic finite state automata (NFA) to find all possible DNA subsequences within some Levenshtein distance. To increase computational efficiency, this NFA is converted to a deterministic finite state automata and inserted into an Aho-Corasick-like data structure to find potential matches across the entire genome.
Source code available for download:
GitHub
Originally published in:
E.K. Pugach, P.A. Richmond, J.G. Azofeifa, R.D. Dowell, L.A. Leinwand
Journal of Molecular and Cellular Cardiology 86:54-61 (2015)
Previous Software

We present a modeling framework aimed at capturing both the positional and temporal behavior of transcriptional regulatory proteins. There is growing evidence that transcriptional regulation is the complex behavior that emerges not solely from the individual components, but rather from their collective behavior, including competition and cooperation. Our framework describes individual regulatory components using generic action oriented descriptions of their biochemical interactions with a DNA sequence. All the possible actions are based on the current state of factors bound to the DNA. We developed a rule builder to automatically generate the complete set of biochemical interaction rules for any given DNA sequence. Off-the-shelf stochastic simulation engines can model the behavior of a system of rules and the resulting changes in the configuration of bound factors can be visualized.
Source code available for download:
SourceForge
Originally published in:
D.A. Knox and R.D. Dowell
IEEE/ACM Transactions on Computational Biology and Bioinformatics May-June 13(3):459-71; ISSN: 1545-5963 (2016)

CodaChrome is a tool for visually exploring how the proteome of a bacterium of interest relates to every other finished bacterial genome in GenBank.
Source code available for download:
Sourceforge
Originally published in:
J. Rokicki, D. Knox, R.D. Dowell, and S.D. Copley
BMC Genomics 15:65 (2014)

GeneProgram uses expression data to simultaneously organize tissues into groups and genes into overlapping programs with consistent temporal behavior, to produce maps of expression programs, which are sorted by generality scores that exploit the automatically learned groupings.
Source code available for download:
Gifford Lab
Originally published in:
G.K. Gerber, R.D. Dowell, T.S. Jaakkola, and D.K. Gifford
PLoS Computational Biology 3(8):e148 (2007)

Consan provides pairwise RNA structural alignment, both unconstrained and constrained on alignment pins.
Source code available for download:
Eddy/Rivas Lab
Originally published in:
R.D. Dowell and S.R. Eddy
BMC Bioinformatics 7:400. (2006)

Joint Binding Deconvolution uses easily obtainable experimental data about chromatin immunoprecipitation (ChIP) to improve the spatial resolution of the transcription factor binding locations inferred from ChIP followed by DNA microarray hybridization (ChIP-Chip) data.
Source code available for download:
Gifford Lab
Originally published in:
Y. Qi, P.A. Rolfe, K.D. MacIsaac, G.K. Gerber, D. Pokholok, J. Zeitlinger, T.W. Danford, R.D. Dowell, E. Fraenkel, T.S. Jaakkola, R.A. Young, and D.K. Gifford
Nature Biotechnology 24(8):963-70. (2006)

Conus is software developed for exploring the consequences of different single sequence Stochastic Context Free Grammar (SCFG) designs in predicting RNA secondary structure.
Source code available for download:
Eddy/Rivas Lab
Originally published in:
R.D. Dowell and S.R. Eddy
BMC Bioinformatics 5:71. (2004)